The Role of Genetic Testing in Clinical Trials
You are here!
Genetic testing can contribute to the development of new treatments for inherited eye diseases in four main ways.
Dr. Edwin Stone
First, it will allow a treatment to be matched to a disease in a rational way. For example, in trials of gene replacement therapies, patients who do not lack the gene that is being therapeutically replaced would not be expected to benefit from the treatment. Inclusion of patients with similar clinical features but a different missing gene will only diminish the apparent utility of the treatment in a trial. Thus, the use of genetic testing to select study participants can increase the power and decrease the cost of a clinical trial.
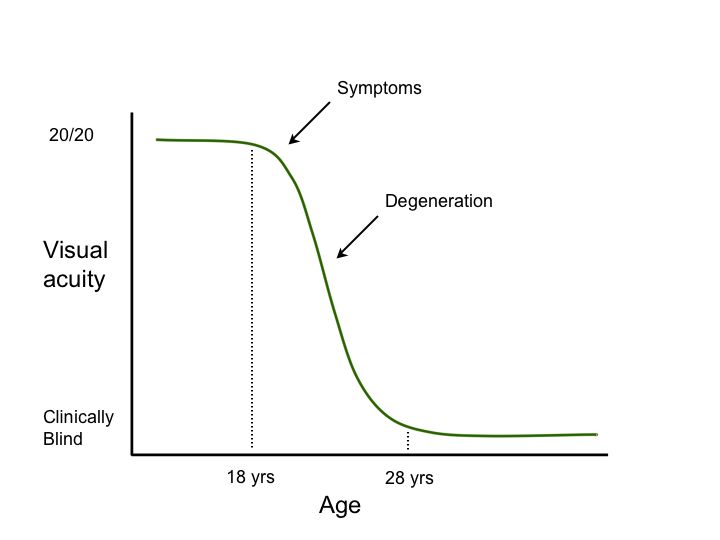
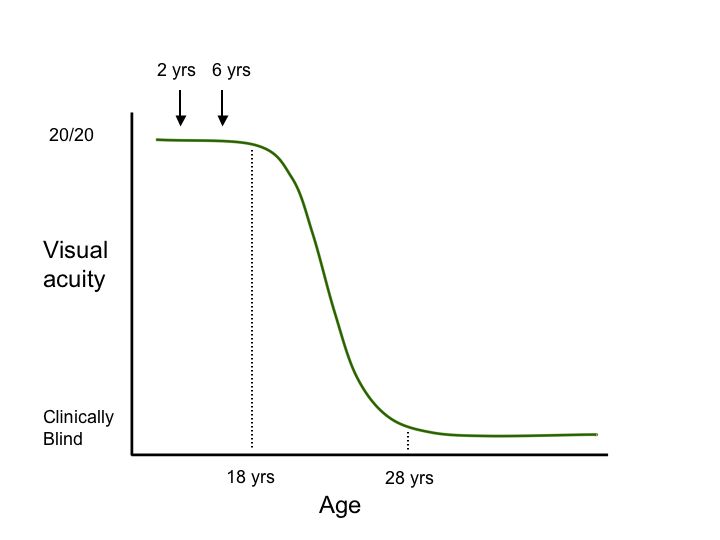
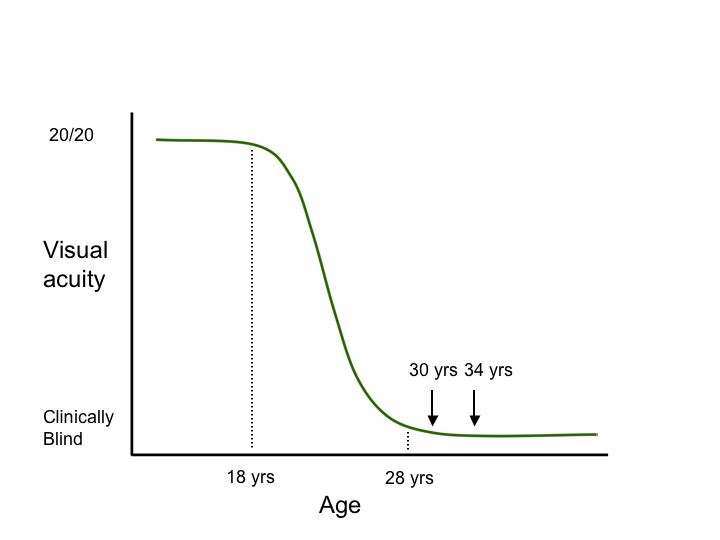
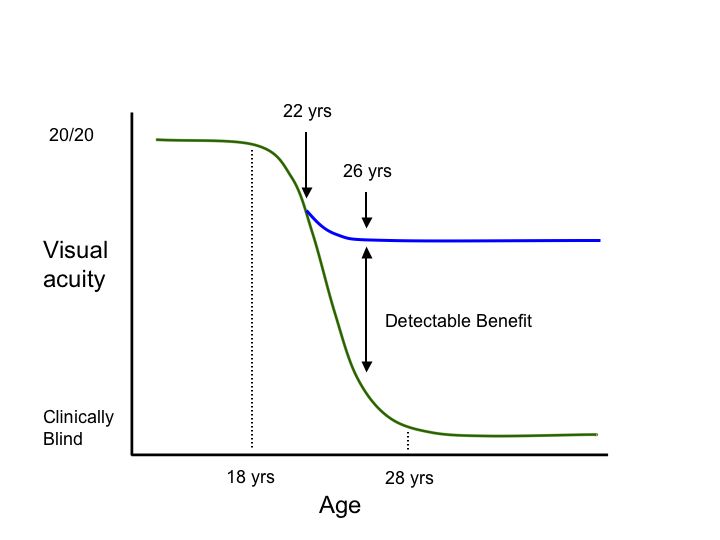
Second, genetic testing can help investigators take advantage of our constantly growing knowledge of the “natural history” of diseases caused by specific genetic variations. Consider the hypothetical degenerative disease shown in figure 1A. In this disease, there is a presymptomatic period from age 0-8, a period of decreasing vision from 8 to 28, and a period of stable but poor vision from age 28 until death. Suppose one planned to conduct a trial of a new cell-death-inhibiting drug over a four year period. If one chose to enroll patients at age 2 and assess the effect of the treatment at age 6 (Figure 1B, arrows), it would be impossible to detect any benefit of the drug because treated individuals and untreated individuals would be expected to be the same during this period. Similarly, if one enrolled patients at age 30 and evaluated them at age 34 (Figure 1C, arrows), there would be no possibility of detecting a benefit because the patients have already experienced all of the cell death they are going to experience by this age and one would not expect any difference between the treated and untreated groups in this study design. That is, even if a drug were fabulously efficacious for arresting the cell death associated with this disease, one would conclude that the drug “had no benefit” if one designed the trial to focus on the youngest or oldest patients affected with the disease. In contrast, if one enrolled patients at age 22 and evaluated them at 26 (Figure 1D, arrows), one would be able to easily detect any beneficial effect of the drug because this is the period during which the cell death occurs in this disease. These gene specific natural history curves can be created by screening large numbers of patients for mutations in known disease genes and then correlating their retrospective clinical data with the disease-causing mutations that are observed.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Third, genetic testing can reduce inter-individual variability in clinical trials. Some genetic variants exhibit a wide range of expressivity within the population (suggesting an important role for other genes and/or the environment in the pathogenesis of the disease) while others have a more predictable behavior. By choosing patients known to have variations with fairly predictable expressivity, one can reduce the variability in the data caused by factors other than the drug being tested and thereby increase the power of a study and decrease its cost.
Fourth, genetic testing can allow presymptomatic treatment to be employed. For many degenerative diseases, by the time the disorder is clinically manifest, more than 50% of the cells at risk for death have already been lost or are so injured that they cannot be rescued by cell protective intervention. Genetic testing of presymptomatic members of families affected with certain diseases (or of the general population if the disease is common enough to warrant the cost of such a maneuver) can allow treatment to be given while the majority of the cells at risk are still healthy enough to respond to the treatment.
Another aspect of genetic testing that is important to consider in the context of a clinical trial of an inherited disease is the specific technique that will be used to perform the testing. The explosion of technology in the past 20 years has provided a number of powerful methodologies that a clinician scientist might consider, but these techniques each have different strengths and weaknesses such that only a few of them would be reasonable to use in any given trial. For example, one might consider the efficiency of each technique in terms of the cost of evaluating a single base pair of DNA for a disease-causing variation. The table below gives the approximate costs of a variety of commonly used approaches.
| Restriction digestion | $3 per base; 3.0 dpb |
| Low density SNP chips | $600 per 10,000 non-contiguous bases; 0.06 dpb |
| DNA sequencing | $12 per 800 contiguous base pairs; 0.015 dpb |
| Medium density SNP chips | $900 per 100,000 non-contiguous bases; 0.009 dpb |
| DHPLC | $3 per 1000 contiguous base pairs; 0.003 dpb |
| SSCP | $0.50 per 200 contiguous base pairs; 0.0025 dpb |
Suppose that one wants to do a trial of a treatment for a disease like malattia leveninese that to our knowledge is caused by only a single mutation (Arg345Trp in the EFEMP1 gene). An allele-specific test based on restriction digestion would cost $3 per patient. Automated DNA sequencing would cost $12 per patient and the use of some solid phase allele detection method would cost as much as $900 per patient. Thus, even though the latter method can evaluate many more base pairs per dollar than DNA sequencing or restriction digestion, this is an empty benefit because one is only interested in one specific base pair in this clinical situation and the added genotypic information can only add irrelevant and potentially misleading information.
A number of inherited eye disorders can be caused by a number of different genes. For example, the phenotype known to clinicians as autosomal recessive retinitis pigmentosa (ARRP) is likely to be caused by over 50 different genes in the human population. Such “locus heterogeneity” creates a very serious problem for a genetic test that evaluates many genes simultaneously. The problem is that the carrier frequency is so high in the population that heterozygous sequence changes in affected individuals have little diagnostic significance. To see why this is so, let’s assume that autosomal recessive RP is caused by 50 different genes which each cause 2% of the ARRP cases in the population of patients that we are testing. Since about 1/6000 individuals are affected by ARRP, approximately 1/300,000 have disease that is caused by a specific one of the 50 genes. The Hardy-Weinberg equation predicts that the carrier frequency of an autosomal recessive disease with this prevalence in the population will be 1/274. However, since there are 50 different genes with this frequency in the population, the cumulative carrier frequency for ARRP is 50/274 or almost 1/5. Thus, one in 5 normal people in the population would be expected to harbor a true disease-causing mutation in one allele of an RP gene. Similarly, one in 5 RP patients would be expected to harbor a true disease-causing mutation in one allele of an RP gene in addition to the mutations that really cause their disease. Thus, one must assume that any heterozygous changes that are observed during multi-locus genetic testing of highly heterogeneous diseases represent this type of irrelevant carrier discovery and one of the true disease-causing mutations.
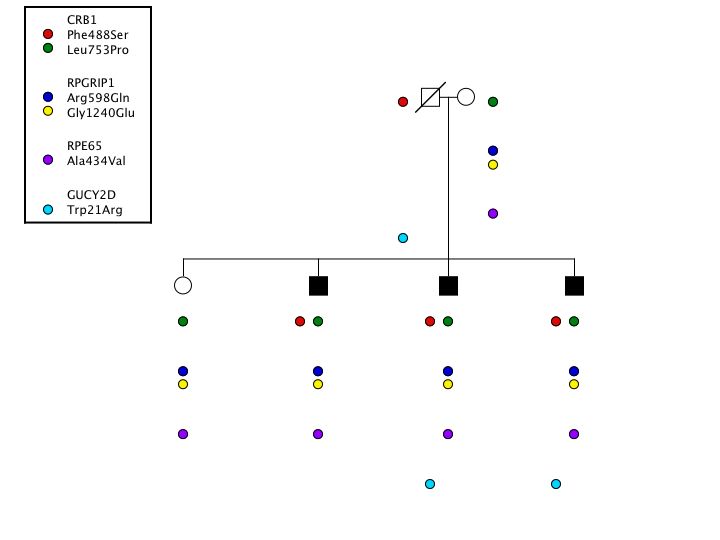
To show that this is more than a theoretical concern, consider the following observations that were made during our screening of a large cohort of patients affected with the genetically heterogeneous condition known as Leber Congenital Amaurosis.(LCA). In this experiment, we screened 8 different genes for mutations in a cohort of 450 probands. In one of these probands, we found six different variations distributed across four genes: CRB1 (Phe488Ser and Leu753Pro); RPGRIP1 (Arg598Gln and Gly124Glu); RPE65 (Ala434Val); and GUCYD2 (Trp21Arg) (Figure 2). Which (if any) of these changes would you feel are responsible for this patient’s disease? What other information would you need to have before you could comfortably answer that question?
{kind=link}
As it turns out, the GUCYD2 variation is clearly a non-disease-causing polymorphism. It is present in 2% of the whole population, which is much too common to cause a disease with a prevalence of 1/50,000. The RPE65 is also clearly a non-disease-causing polymorphism because it is present in 11% of the African American population. If one had used an entirely Caucasian control group, one might have erroneously concluded that this was a disease-causing variant. Thus, it is very important to screen a large control group with the same ethnic composition as the patient population being tested to avoid this type of error. Finally, the RPGRIP mutations were both found to lie on the maternal allele. This could only be detected by examining the parents of the proband. Having eliminated the RPGRIP1, RPE65 and GUCYD variants, and demonstrating that the two CRB1 variants were each inherited from a different parent, one can deduce that the latter two variations are the most likely cause of this individual’s disease.
In conclusion, genetic testing will play a very important role in clinical trials of novel treatments for inherited eye diseases both by identifying individuals who are most likely to benefit from the therapy and by allowing the therapy to be evaluated at the point in the natural history of the disease that is most likely to result in a detectable difference between treated and untreated patients. No genetic testing platform is ideal in all respects. However, in general, the more focused a molecular hypothesis can be (on clinical grounds) before genetic testing is performed, and the fewer genes that need to be screened to evaluate this hypothesis, the less expensive the test will be and the less likely one will be misled by a variation in a gene other than the one that is truly causing the patient’s disease.
« Return to the Clinical Trials FAQs